
Journal of Clinical Review & Case Reports(JCRC)
ISSN: 2573-9565 | DOI: 10.33140/JCRC
Impact Factor: 1.823


Case Report - (2023) Volume 8, Issue 10
Renal Salt Wasting: Earliest Manifestation of Rhabdomyolysis, Supreet/Mehand-ru Syndrome
2Department of Nephrology and Hypertension, Cleveland Clinic Lerner College of Medicine of Case Western Reserve University, Glickman Urological & Kidney Institute Cleveland Clinic, Cleveland Ohi, USA
Received Date: Sep 19, 2023 / Accepted Date: Sep 29, 2023 / Published Date: Oct 15, 2023
Copyright: �©2023 Supreet Kaur, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original authors and source are credited
Citation: Kaur, S., Masud, A., Singh, P.P., Rezkalla, K., Efobi, R., et al. (2023). Renal Salt Wasting: Earliest Manifestation of Rhab-domyolysis, Supreet/Mehandru Syndrome. J Clin Rev Case Rep, 8(10), 239-247.
Abstract
Acute kidney injury is a feared and common complication of rhabdomyolysis, occurring in 10-40% of hospitalized patients with mortality rate as high as 59% in critically ill patients. However, there is a lack of information in the literature on renal salt wasting in rhabdomyolysis. We report 3 cases of rhabdomyolysis who developed renal salt wasting in presence of normal glomerular filtration rate. To the best of our knowledge, this is the first case report in humans displaying this phenomenon. Authors noted that all the three patients had urine pH of over 7.0. Literature reveals, patients with rhabdomyolysis and urine pH<6.5 develop acute kidney injury whereas urine pH>7.0 experienced renal salt wasting in presence of normal glomerular filtration rate. All 3 cases, previously not known to have hypertension, were noted to have elevated blood pressure during acute phase of rhabdomyolysis. Animal studies have shown that increase in blood pressure causes increase in renal interstitial pressures that results in naturesis and diuresis. All 3 cases had increased levels of plasma renin activity and aldosterone. There is a direct proportionality between increased blood pressure and naturesis. Clinicians should be aware of salt wasting as an early renal manifestation in patients with rhabdomyolysis.
Keywords
Rhabdomyolysis, Renal salt wasting, Acute kidney failure, Urine pH, Naturesis
Introduction
Literature has exhibited significant relationship between urine pH and AKI. In a study conducted by Mehandru et al., majority of the patients with urine pH of 7.0 had normal GFR [1]. Kidney micro-puncture studies in animals have shown, unexpectedly low intratubular pressures after exposure to myoglobin. Myoglobinuria reduced intratubular pressure resulting in reduced sodium and water reabsorption in proximal convoluted tubule results in renal salt wasting (RSW). Protective role of alkaline urine has also been demonstrated in studies where animals were injected with myoglobin or hemoglobin. In rat studies, direct toxic effects of myoglobin or its degradation products, pigment cast formation with tubular obstruction, and renal vasoconstriction with hypoxic tubular damage have been blamed for myoglobin nephrotoxicity [2-4] (Figure 1).
Urine pH>7.0 results in significant histological renal protection, tubular necrosis being either totally absent or minimal in its existent [2]. Myoglobinuria has been reported to result in AKI by direct renal tubular toxicity, vasoconstriction, and uric acid cast formation. AKI has been found in majority of patients with urine pH7.0, however all patients with urine pH>7.0 had urine salt wasting with normal GFR.
AKI develops in 10-40% patients with severe rhabdomyolysis. No single factor including creatinine phosphokinase (CPK), serum creatinine, potassium (K+ ), calcium (Ca++), or myoglobin in urine accurately predicts AKI in patients with rhabdomyolysis [5].
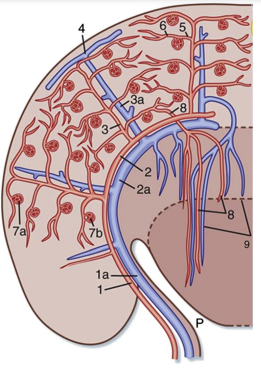
Figure 1: This is an illustration of the vascular system of the human kidney. 1, interlobar arteries; 1a, interlobar vein; 2, arcuate arteries; 2a, arcuate veins; 3, interlobular arteries; 3a, interlobular veins; 4, stellate vein; 5, afferent arterioles; 6, efferent arterioles; 7a, 7b, glomerular capillary networks; 8, descending vasa recta; 9, ascending vasa recta. Taken from Koeppen, B.A., Stanton, B.A., 2010. Berne and Levy Physiology, sixth ed. Elsevier, Chapter 32 (Elements of Renal Function). Figure 32-2, p. 559. (REF: EJ Johns & AF Ahmeda)
Case Report
All three patients reported in this study presented with rhabdomyolysis and exhibited increased urinary sodium excretion. These patients also experienced hypertension during the acute phase of rhabdomyolysis that resolved upon reduction in CPK levels. Renal functions remained stable in all patients during the course of rhabdomyolysis. Aldosterone and renin levels were also elevated during this time. Urine pH in all three patients remained above 7.0, thus avoiding acute renal injury. Myoglobin in urine varied in the patients in the current study.
Case # 1
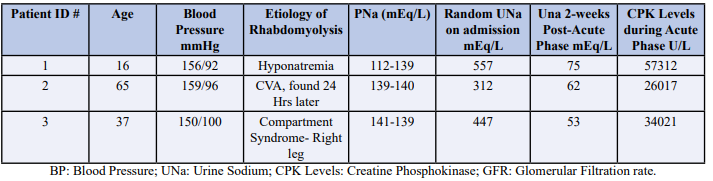
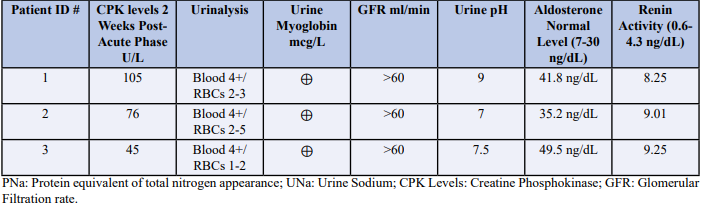
A 16-year-old male was admitted with generalized body aches, weakness, and high blood pressure in the range of 156/92 mmHg with heart rate 91 BPM, and normal body temperature 98.5ºF. Patient was employed as a lifeguard on the beach with a day temperature 95ºF and humidity 74%. Patient had a normal physical examination at admittance with CPK level at 57,312 (U/L). The plasma sodium (Na) level was low at 112 ng/dl, potassium (K) level at 4.2 mEq/L, bicarb (HCO3) level at 22 mEq/L and Urine output was 1562 cc over the period of 24 hrs. Urine sodium was at 557 mEq/L and Serum creatinine at 1.10 ng/dl. Hemoglobin was in normal range of 13.2 g/dl. Urinalysis showed 4+ blood, RBCs count 2-3, with urine pH at 9.0. Additionally, Urine myoglobin was also present. Aldosterone was noted 41.8 ng/dL and plasma renin activity was 8.25 ng/dL. Patient was given intravenous normal saline with improvement in overall condition.
1-week post admittance patient was asymptomatic with improved Blood pressure (148/90 mmHg) and heart rate 91 BPM. The CPK levels were also greatly reduced 7692 U/L and Plasma Na (132 ng/ dl), K (4.0 mEq/L) and HCO3 (23 mEq/L) were greatly improved. Na level in urine was decreased to 210 mEq/L and Urinalysis revealed 3+ blood, 0-2 RBCs, pH 7.0, suggesting improvement with patient. Urine myoglobin negative. Aldosterone was noted 33.9 ng/dL and plasma renin activity was 7.02 ng/dL. As the condition of patient improved, he was kept on regular diet, IV fluids discontinued.
At 2-week post admittance, patient remained asymptomatic with BP (124/86 mmHg) and HR (72 BPM) improving towards normal range. The CPK levels (432 U/L), plasma Na 138 mEq/L, K 4.3 mEq/L, HCO3 24 mEq/L improved. Urine Na level was also in normal range of 76 mEq/L. Urinalysis revealed +blood, no RBCs, and pH 7.5. Urine myoglobin was negative Aldosterone was noted 3.2 ng/dL and plasma renin activity was 4.1 ng/dL. Patient was on regular diet at home, back to school.
On the 3-week mark, Patient remained asymptomatic with BP (122/74 mmHg), HR (70 BPM), and T 97.0ºF in normal range. CPK levels were at 92 U/L with Plasma Na 139 mEq/L, K 3.9 mEq/L, HCO3 23 mEq/L being normal. Urine Na was also normal at 56. Urinalysis revealed no blood, no RBCs, pH 7.0 with negative urine myoglobin. Aldosterone was noted 3.2 ng/dL and plasma renin activity was 1.3 ng/dL. Patient remains stable without any issues.
Case # 2
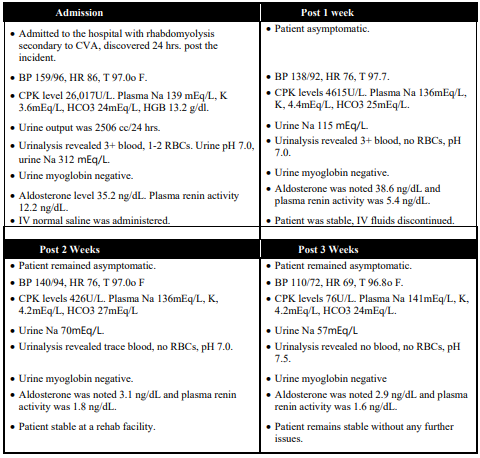
A 65-year-old female was admitted to the hospital with rhabdomyolysis secondary to CVA, which was discovered 24 hrs. post the incident. She had high blood pressure in the range of 159/96 mmHg with heart rate 86 BPM, and normal body temperature 97ºF. The patient had CPK level at 26,017 U/L and plasma Na level at 139 mEq/L, K level at 3.6 mEq/L, and HCO3 was at 24 mEq/L. Her urine output was 2506 cc over the period of 24 hrs. Urine sodium was at 312 mEq/L with Hemoglobin in normal range of 13.2 g/dl. Urinalysis had 3+ blood, RBCs counts at 1-2, with urine pH normal at 7.0. Patient had no Urine myoglobin with aldosterone level at 35.2 ng/dL and plasma renin activity was 12.2 ng/dL. Patient was given intravenous normal saline with improvement in overall condition.
At the 1-week post admittance, patient improved a lot. She was asymptomatic with improved BP (138/92 mmHg) and HR at 76 BPM. The CPK levels were improved to 4615 U/L and Plasma Na (136 mEq/L), K (4.4 mEq/L) and HCO3 (25 mEq/L) were greatly improved. Na level in urine decreased to 115 mEq/L with urinalysis revealing 3+ blood, no RBCs, pH at 7.0. Urine myoglobin was negative. Aldosterone was at 38.6 ng/dL and plasma renin activity was 5.4 ng/dL. Patient was in stable condition and IV fluids were discontinued.
Patient remained asymptomatic, at 2-week post admittance, with BP (122/74 mmHg) and HR (70 BPM) in range. The CPK levels (426 U/L), plasma Na 136 mEq/L, K 4.2 mEq/L, HCO3 23 mEq/L came into normal range. Urinalysis showed trace blood, no RBCs, and urine pH at7 with no urine myoglobin. Aldosterone was at 3.1 ng/dL and plasma renin activity was 1.8 ng/dL. Patient was in stable condition in rehab facility.
Upon 3rd-week mark, patient was asymptomatic with BP (110/72 mmHg), HR (69 BPM), and T 96.8ºF in normal range. CPK levels in normal range at 76 and Plasma Na 141 mEq/L, K 4.2 mEq/L, HCO3 24 mEq/L were in normal range. Urine Na was also normal at 57 mEq/L. Urinalysis revealed no blood, no RBCs, pH 7.5 with negative urine myoglobin. Aldosterone was at 2.9 ng/dL with plasma renin activity was 1.6 ng/dL. There were no noted issues with patient remains in stable condition.
Case # 3
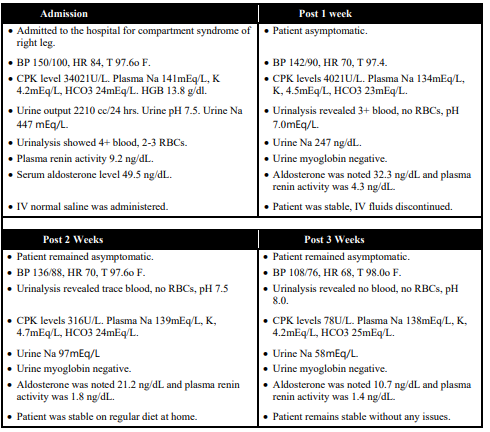
37-year-old male was admitted to hospital with compartment syndrome of right leg with high blood pressure at 150/100 mmHg and heart rate at 84 BPM, and normal body temperature 97.6ºF. Patient had a high CPK level at 34,021 U/L with plasma Na level at 141 mEq/L, K at 4.2 mEq/L, and HCO3 at 24 mEq/L. Urine output was 2210 cc over the period of 24 hrs with urine sodium at 447 mEq/L and urine pH at 7.5. Patient’s urinalysis depicted 4+ blood and RBCs count at 2-3. Patient’s aldosterone level was noted at 49.5 ng/dL and plasma renin activity at 9.2 ng/dL. Patient was administered IV normal saline.
At the 1-week post admittance, patient was asymptomatic with BP (142/90 mmHg) and heart rate 70 BPM. The CPK levels were improved at 4021 with Plasma Na (134 mEq/L), K (4.5 mEq/L) and HCO3 (23 mEq/L). Urine Na level were at 247 mEq/L with Urinalysis revealed 3+ blood, no RBCs, pH 7.0. Urine myoglobin was negative, and aldosterone was noted 32.3 ng/dL and plasma renin activity was 4.3 ng/dL. Patient was stable and IV fluids discontinued.
On the 2-week post admittance point, patient remained asymptomatic with improvement in BP (136/88 mmHg) and HR (70 BPM). The CPK levels were at 316 U/L, plasma Na 139 mEq/L, K 4.7 mEq/L, HCO3 24 mEq/L were significantly improved. Urine Na level was in range of 76 mEq/L with urinalysis revealing trace blood, no RBCs, and pH 7.5. Urine myoglobin was negative Aldosterone was noted 21.2 ng/dL and plasma renin activity was 1.8 ng/dL. Patient was put on regular diet at home.
3-week post admittance to hospital, patient was asymptomatic with BP (108/76 mmHg), HR (76 BPM), and T 98.0ºF in normal range. CPK levels were at 78 U/L and Plasma Na level at 138 mEq/L, K 4.2 mEq/L, HCO3 25 mEq/L being in normal range. Urine Na was at 58 with urinalysis revealing no blood, no RBCs, pH 8.0 with negative urine myoglobin. Aldosterone was noted 10.7 ng/dL and plasma renin activity was 1.4 ng/dL. Patient was stable at this point without any issues.
Case# 1: Male/16 years

Case# 2: Female/65 years
Case# 3: Male/37 years
Discussion
Early reviews of mechanism of renal dysfunction in rhabdomyolysis (RM) noted myoglobin as an inert participant causing tubular obstruction and secondary renal dysfunction. Tubular obstruction has been inferred by the presence of tubular dilatation, which has been found in histological section. Micro-puncture renal studies show that intratubular pressures are unexpectedly low, which results in reduced absorption of sodium and water, resulting in naturesis and diuresis. Under physiological environment with urine pH>7.0, myoglobin induced renal dysfunction does not rise to the level of AKI but results in RSW. In our present study, myoglobin induced AKI under acidic urine (pH7.0 RSW is noted. In a study conducted by Mehandru et al., significance relationship between urine pH and AKI was noted, majority of the patients in this study with urine pH of 7.0 had normal GFR [1].
Blood pressure (BP) in all 3 patients in this study, not known be hypertensive, was elevated. Plasma renin activity (PRA) and aldosterone were also elevated in all 3 cases. This is consistent with Goldsmith & Hicks publication, where authors described 2 cases with acute RM developing acute hypertension with elevated PRA and aldosterone. Urine sodium was, however, not obtained in that study [6]. Further studies may elicit cause of hypertension in patients with rhabdomyolysis. Mechanism in which sodium and water excretion is impacted by change in BP is very different than that of renal blood flow. In fact, there is direct proportionality between increase in BP and the rise in urine sodium excretion [7]. Removal of renal capsule in animals eliminates this naturetic effect (Figure 2).
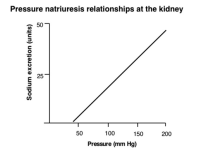
Figure 2: Relationship between blood pressure and sodium excretion (REF: EJ Johns & AF Ahmeda)
The underlying mechanism of this phenomenon has been debated for some time but has not been resolved. Measurements have clearly shown that as renal perfusion pressures rise, interstitial pressure also increases [8]. Thus, it would seem that the rise in hydrostatic pressure is transmitted along with the arcuate and intralobular arteries into the intrestitium. Clearly, the autoregulatory vasoconstrictor response at the arteriolar resistance vessels prevents the pressure rise from reaching the glomerulus, as GFR remains stable. However, the hydrostatic pressure is transmitted into the intrestitium, it does not have the potential of increasing hydrostatic pressure within the peritubular capillaries in the cortex that surround the proximal tubules [7]. This increase in capillary pressure would depress the reabsorption of fluids in the proximal tubules as it is an important component of the sterling forces and the result would be an increase in sodium and water excretion [8]. This phenomenon explains pressure naturesis, although the underlying mechanisms remain unexplained.
Infusion of myoglobin (MB), in animals, causes renal vasoconstriction in a pH dependent manner. Studies in isolated perfused kidney demonstrated that MB causes little change in profuse rate flow at physiological pH, but acidic pH level induces profound vasoconstriction together with a reduction in GFR and tubular reabsorption of sodium. This may apply to our three cases presented herein. All three patients in our publication had rhabdomyolysis, urine pH of 7-9, normal GFR, and had renal salt wasting. The process of intratubular cast formation and increase in renal salt wasting is depicted in figure 3 as mentioned by Mehandru et al. [1] (Figure 3). This phenomena in humans has not been described in literature. We believe, based upon the above study acidic pH results in AKI by vasoconstriction and no AKI in subjects with urine pH over 7.0. We also believe that RSW is related to:
a) Reduced intratubular pressure resulting in reduced sodium and water reabsorption in proximal convoluted tubules.
b) Alkaline urine with pH>7.0 results in RSW rather than AKI.
c) Hypertension (All 3 patients had elevated BP during acute episode of rhabdomyolysis) causes increased sodium and water excretion. There is a direct proportionality between increased BP and rise of urine sodium excretion.
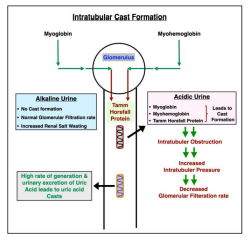
Figure 3: Intratubular Cast Formation and Increase in Renal Salt Wasting (Mehandru et al.)
The protective role an alkaline urine plays against the development of AKI has been demonstrated in animals injected with hemoglobin or myoglobin. Urine alkalization could improve the solubility of urates, the product of nucleic acid breakdown during tissue damage [7]. Intratubular and renal intracellular pH could also affect the fate of heme proteins within the kidney: enhanced renal retention of myoglobin was noticed in acidotic animal [10].
Systemic acidosis was shown to correlate with kidney function deterioration in patients with rhabdomyolysis [11]. This may indicate the presence of hemodynamic instability or volume depletion with tissue hypoperfusion, rather than being an independent contributing factor for the development of myoglobinuric nephropathy [4].
Systemic acidosis has been suggested as a risk factor in development of pigment nephropathy [11-13] because of enhanced renal retention of M (myoglobin) [2,10], augmented cast formation [2], and increased release of acid [14]. Since urine acidification takes place at the distal nephron, it is conceivable that heme release may especially be pronounced within the medullary structures. This, in turn, could inactivate local nitrovasodilation, inducing vasoconstriction, exacerbation of regional physiological oxygen insufficiency, and outer medullary hypoxic injury [4]. There may be some potential protective properties of acidosis. Acidosis may confer protection by reduction of oxygenhemoglobin affinity, enhancing oxygen extraction by tissue [15].
Acidosis may also intensify nitric oxide synthesis [16] and overcome nitric oxide trapping by heme pigment [17]. In addition, acidosis may reduce vasoconstrictive response to angiotensin II and to α2 sympathetic agonists [18-20]. Acidosis also reduces left ventricular contractility and systemic vascular resistance [21]. The protective role of an alkaline urine against development of AKI has been demonstrated in animals injected with hemoglobin or myoglobin.
Myoglobin induces renal injury by mechanisms that remain incompletely defined. Acidosis has been suggested as an important factor in myoglobinuric renal failure, and urine alkalization is routinely recommended for patients for prevention of AKI.
In isolated rat kidney at physiological urine pH (urine pH>7.0), myoglobin at concentrations of 20-250 mg/dL minimally effected renal perfusion flow, GFR and tubular sodium reabsorption. Tubular sodium reabsorption (TRNa) by contrast, in serum pH<7.0 results in myoglobin induced vasoconstriction, reduced GFR, and TRNa and increased hypoxic injury to medullary thick ascending limbs [4].
It appears that variety of physiological factors, in addition to urine pH and solute excretion, are critical determinants of myoglobin nephrotoxicity which appears to exert themselves after initial phase of renal myoglobin deposition. One such factor might be the rate of myoglobin cast dissolution/excretion. A shorter period of cast retention should shorten proximal tubular urinary status, thereby decreasing proximal tubule cell myoglobin uptake and hence, limiting necrosis [22].
The importance in recognizing the inability in the maintenance phase result implies that therapeutic intervention might abort myoglobin AKI even after the phase of acute renal myoglobin deposition has occurred.
Aciduria promotes myoglobinuric renal injury by acutely trapping myoglobin (M) within the kidney, not by causing hematin formation. Sodium bicarb (HCO3) protects by increasing M solubility by providing non-absorbable solute, both facilitating its excretion. Fe-stimulated hydroxyl radical formation does not appear to be necessary for M to cause renal damage [2].
It has been generally accepted that M can exert a nephrotoxic potential when excreted under aciduric but not alkalinuric condition (2,10,13,23-25). The prevailing explanation for this observation is that during aciduria (urine pH<5.6) the Fecontaining porphyrin ring is split from M, producing ferrihemate (hematin) [10,27,28]. The latter is believed to have a direct nephrotoxic effect on proximal tubular cells (PTCs). It is possible that the protective effect of bicarbonaturia is related not only to prevention of hematin formation but rather to HCO3 related solute diuresis.
The most prominent histological abnormality noted in ammonium chloride (NH4Cl)-treated rats [3] was patchy proximal tubular necrosis, almost totally confined to the outer cortex. However, intra-peritoneal potassium bicarbonate (KHCo3) and potassium sulfate (K2So4) treatment group showed significant histological protection, necrosis being either totally absent to minimal in its extent. Only minimal to mild medullary vascular congestion and cast deposition were noted [3].
Conclusion
Myoglobinuria has been reported to result in AKI by direct renal tubular toxicity, vasoconstriction, and uric acid cast formation. This is the first study in humans, to the best of our knowledge, describing development of RSW (AKI in majority of cases with urine pH7.0. Myoglobin reduces intratubular pressure resulting in reduced sodium and water reabsorption in proximal convoluted tubules causing RSW. Hypertension, found in all 3 cases resulted in increased sodium and water excretion. There is a direct proportionality between elevated BP and rise in excretion of urine sodium.
Conflict of Interest Statement
None of the authors declare any conflict of interest.
Acknowledgement
None.
Funding Sources
None.
Informed Consent
Informed consents were obtained from the patients.
Data Availability
The data supporting the findings of this study are available from the corresponding author upon reasonable request.
References
1. Mehandru, S., Kaur, S., Masud, A., Khan, Q., Rezkalla, K., et al. (2023). Significance of Urine pH in Predicting Renal Outcomes for Patients with Rhabdomyolysis. J Clin Rev Case Rep, 8(8), 149-156
2. Zager, R.A. (1989). Studies of mechanisms and protective maneuvers in myoglobinuric acute renal injury. Lab Invest, 60(5), 619-629.
3. Zager, R.A., & Gamelin, L.M. (1989). Pathogenic mechanisms in experimental hemoglobinuric acute renal failure. Am J Physiol, 256, F446.
4. Heyman, S.N., Rosen, S., Fuchs, S., Epstein, F.H., & Brezis, M. (1996). Myoglobinuric acute renal failure in the rat: a role for medullary hypoperfusion, hypoxia, and tubular obstruction. J Am Soc Nephrol, 7(7), 1066-1074.
5. Esposito, P., Estienne, L., Serpieri, N., et al. (2018). Rhabdomyolysis-Associated Acute Kidney Injury. American Journal of Kidney Disease, 2018, 71(6).
6. Goldsmith, B.M., Hicks, J.M. (1985). Rhabdomyolysis: two pediatric case reports. Clin Chem, 31(2), 314-317.
7. Johns, E., & Ahmeda, A. (2014). Renal Circulation. Elsevier.
8. Garcia-Estan, J., & Roman, R.J. (1989). Role of renal interstitial hydrostatic pressure in the pressure diuresis response. Am J Physiol, 256, F63-F70.
9. Bank, N. (1991). Better as: Acid-base balance and acute renal failure. Miner Electrolyte Metab, 17, 116-123.
10. Bywaters, E.G.L. & Stead, J.K (1944). The production of renal failure following injection of solutions containing myohaemoglobin. Quart J Exp Physiol, 33, 53.
11. Baker, S.L. & Dodds, E.C. (1925). Obstruction of Renal Tubules During the Excretion of Hemoglobin. Br J Exp Pathol, 6, 247-260.
12. Muckart, D.J.J., Moodley, M., Naidu, A.G., Reddy, A.D.R., & Meineke, K.R. (1992). Prediction of Acute Renal Failure Following Soft-Tissue Injury Using the Venous Bicarbonate Concentration. J Trauma, 33, 813-817.
13. Perri, G.C., & Gorini, P. (1952). Uremia in the rabbit after injection of crystalline myoglobin. Br J Exp Pathol, 33, 440.
14. Litwin, M.S., Walter, C.W., & Jackson, N (1960). Experimental production of acute renal tubular necrosis. I. The role of gram-negative bacteremia II. The toxicity of acid hematin. III. Acid hematin-the etiologic pigment. Ann Surg, 152, 1010-1025.
15. West, J.B. (1988) Respiratory physiology. Ventilation, blood flow and gas exchange; in Nadel lA, Murray JF (eds): Textbook of Respiratory Medicine. Philadelphia, Saunders, 1988, 47-84.
16. Baud, L., Boutard, V., & Fouqueray, B. (1995) Modulation of nitric oxide synthase activity in rat macrophages by intracellular pH (pH;). J Am Soc Nephrol, 6, 822.
17. Heyman, S.N., Greenbaum, S., Shina, A., Rosen, S., & Brezis, M. (1997). MYOGLOBINURIC acute renal failure in the rat: A Role for Acidosis. Experimental Nephrology, 5, 210-216.
18. Chen, L.Q., Shepherd, A.P. (1991). Role of H+ and alpha 2 receptors in escape from sympathetic vasoconstriction. Am J Physicl 26I, H868- H87J.
19. Tateishi, 1., & Faber, J.E. (1995). Inhibition of arteriole alpha 2- but not alpha l-adrenoceptor constriction by acidosis and hypoxia in vitro. Am 1 PhysioI 268, H2068-H2076.
20. Rose, C.E. Jr., Walker, S.R., Erickson, A., Kaiser, D.L., Carey, R.M., et al. (1982). Renal and cardiovascular responses to acute hypercapnic acidosis in conscious dogs: Role of renin-angiotensin. J Cardiovasc Phannacol, 4, 676- 687.
21. Walley, K.R., Lewis, T.H., & Wood, L.D. (1990). Acute respiratory acidosis decreases left ventricular contractility but increases cardiac output in dogs. Circ Res, 67, 628-635.
22. Zager, R.A., & Gamelin, L.M. (1989). Pathogenic mechanisms in experimental hemoglobinuric acute renal failure. Am J Physiol, 256, F446.
23. Dubrow, A., & Flammenbaum, W. (1988). Acute renal failure associated with myoglobinuria and hemoglobinuria. In Acute Renal Failure, Chapter 10, Ed 2, edited by Brenner BM, Lazarus JM, p 279. New York, Churchill Livingstone.
24. Garcia, G., Snider, T., & Feldman, M. (1981). Nephrotoxicity of myoglobin in the rat: relative importance of urine pH and prior dehydration (abstr). Kidney Int, 19, 200.
25. Kagen, K.L. (1971). Myoglobinemia and myoglobinuria in myositis syndrome. Arthritis Rheum, 14, 457.
26. Ron, D., Taitelman, M.D., Michaelson, M.D., Gad, B.J., Bursztein, E., et al. (1984). Prevention of acute renal failure in traumatic rhabdomyolysis. Arch Int Med, 144, 277.
27. Brezis, M., Rosen, S., & Epstein, F.H. (1986). Acute Renal Failure. In The Kidney Chapter 19. Ed 3, edited by Brenner BM, rector FD, P 743-Philadelphia W. B. Saunders, 1986.
28. Bunn, H.F., & Janell, J.H. (1966). Exchange of heme among hemoglobin molecules. Proc Natl Acad Sci USA, 56: 974.