
Journal of Clinical Review & Case Reports(JCRC)
ISSN: 2573-9565 | DOI: 10.33140/JCRC
Impact Factor: 1.823


Case Report - (2022) Volume 7, Issue 11
Extensive glandular and neuroendocrine differentiation of a malignant peripheral nerve sheath tumor in a neurofibromatosis-1 patient
2Department of Orthopedic Surgery, Children’s Hospital Los Angeles and Keck School of Medicine of USC, USA
3Department of Radiology, Children’s Hospital Los Angeles, USA
Received Date: Nov 16, 2022 / Accepted Date: Nov 18, 2022 / Published Date: Nov 25, 2022
Abstract
The histologic diagnosis of a malignant peripheral nerve sheath tumor (MPNST) is complex with no defining biomarkers or generally accepted diagnostic criteria, but based on whether it is identified arising from a peripheral nerve, arising from a benign nerve sheath tumor, in the setting of neurofibroma or when its constellation of features suggest a Schwann-cell differentiation [1- 3]. Further complicating the histologic diagnosis is the small subset of these tumors with divergent differentiation demonstrating various heterologous elements [4]. Divergent differentiation presenting in a glandular form in MPNST is particularly rare, though has been described, usually in context of Neurofibromatosis I (NF1) patients, as discrete localized areas occasionally associated with neuroendocrine features [3,5,6]. Being aware of the possible extent of variable histologic features is critical for diagnosis, particularly if only a needle core biopsy is provided. We present a case of MPNST arising in a 20-year-old woman with NF1 found to have an unusual extensive glandular and neuroendocrine divergent differentiation comprising more the 75% of the tumor, arising from a plexiform neurofibroma. The patient had a prior MPNST resected without evidence of divergent differentiation, and then a subsequent metastasis to the base of the skull with similar glandular histology following our presenting case. To our knowledge, such extensive glandular and neuroendocrine differentiation has not been previously described in these tumors.
Keywords
MPNST, Glandular, Neuroendocrine, Divergent, NF1.
Introduction
Malignant peripheral nerve sheath tumors (MPNST) have a reported incidence of 0.001% and 4.6%, in the general clinic population and Neurofibromatosis 1 (NF1) population respectively, with a reported 5-year disease-specific survival rate of 39%. While approximately half of MPNSTs occur in Neurofibromatosis 1 (NF1) patients, there is no significant correlation between the disease status and MPNST behavior [1,7,8].
Histologic diagnosis is complex as MPNST have been defined as loosely as a sarcoma arising from a peripheral nerve or neurofibroma to more stringent criteria based on origin and constellation of histologic features [1,3]. Difficulty constructing defining criteria arises from the tumors variable histologic appearance and lack of definitive markers [2]. Some cases require differentiation from the benign counterpart neurofibroma, in which case the loss of H3K27me3 immunohistochemical staining has been found to delineate towards an MPNST and inferior prognosis. However, the marker does not differentiate MPNST from other high grade sarcomas such as synovial sarcomas or dermatofibrosarcoma protuberans which have also been found to demonstrate loss of H3K27me3 [9].
The diagnosis is even further complicated as a small subset have been found to demonstrate divergent differentiation, with mesenchymal or epithelial components [4]. Mesenchymal heterologous components including skeletal muscle, osteoid and chondroid tissues, are the most commonly reported. Though rare, the presence of epithelial differentiation is well established particularly in association with the NF1 patient population. In a recent review of glandular differentiation, one of the rarest presentations, the spectrum of cytology ranged from benign to malignant and was described as discrete or localized to a few areas of the tumor. While glandular differentiation complicates the histologic diagnosis, there is no definitive correlation to prognosis [5].
Presenting Case
We present a case of a 20-year-old woman with a complex history of NF1 complicated by developmental delay, seizure disorder, pelvic rhabdomyosarcoma, giant cell granuloma of the mandible and MPNST of the right upper thigh, presenting with an enlarging mass in the right upper arm. She reported increasing pain in the right arm over a course of several weeks, and was noted to have decreased use of her right extremity compared to baseline by a caretaker. Clinical examination revealed a palpable mass in the medial portion of the right upper arm, marked thenar atrophy of the right hand, and generalized right upper extremity weakness. Magnetic resonance imaging (MRI) was performed revealing a 3.1x2.6x11.9 cm hyperintense T2-weighted multilobulated lesion within the biceps brachia, coracobrachialis and medial head of the triceps brachii along the median nerve (Figure 1A). The expansile lesion extended from proximal of the biceps brachia to distal of the elbow joint. There was no evidence of cortical involvement of the humerus, and post contrast imaging showed peripheral and central heterogenous enhancement. Given the clinical history, the radiologic findings corresponded to a diagnosis of MPNST in the setting of NF1.
The patient underwent radical resection of the lesion with partial excision of the median nerve and subsequent reconstruction. Per the operation note, the mass was easily identified without evidence of direct extension into the muscle. The brachial artery was dissected free from the tumor without breech of the tumor capsule. A pathology intraoperative consultation was performed at the time of surgery without evidence of tumor at either the proximal or distal resection margins.
The specimen was received by pathology and consisted of a tanlight brown, elongated and encapsulated soft tissue, with proximal and distal resection margins indicated. It measured 10.5 cm proximal to distal and had a diameter ranging from 0.6 to 3.2 cm. The specimen was inked and serially sectioned to reveal tan-white to light yellow, nodular and slightly mucinous cut surface with a focal area of hemorrhage and necrosis.
Histologic sections demonstrated a lobular lesion, with smooth well-demarcated pseudo encapsulated borders, comprised of a glandular proliferation with mucinous matrix, spindle cells arranged in long fascicles and nerve bundles within fibrous stroma. The lobules of glandular proliferations comprise the majority of the lesion, over 75% examined (Figure 1B), and range from low grade gland structures lined by cuboidal cells demonstrating medium sized ovoid nuclei, fine chromatin, indistinct nucleoli and moderate amounts of apical cytoplasm and interspersed goblet cells to malignant appearing back to back glands with overlapping columnar cells demonstrating moderate pleomorphism, enlarged nuclei, irregular nuclear borders, open to coarse granular chromatin and high nuclear to cytoplasmic ratios. The majority of the glandular proliferation arises in the mucinous matrix with occasional mucin pools, but focally glands are seen extending into spindled cell areas (Figure 1C). The fascicles of spindled cells are moderately to highly cellular, comprised of mildly pleomorphic cells with elongated, small to medium sized nuclei with tapered ends, coarse chromatin and poorly demarcated cellular borders. Mitotic figures and apoptosis are frequently found in the glandular and spindled cell regions of the tumor. Foci of necrosis are present throughout. Focal small solid clusters and trabeculae of cells, within the regions of the glandular proliferations, demonstrate small ovoid nuclei with fine and coarse chromatin and moderate amounts of pale eosinophilic to clear cytoplasm (Figure 1D). The overall lobular architecture including the arrangement of the adjacent nerve bundles gives the overall appearance of the tumor arising from a pre-existing plexiform neurofibroma.
Immunohistochemical stains were performed in this case. Epithelial marker Pan Keratin (AE1/AE3) was strongly positive in the glandular epithelial cells. Neural marker S100 was rare in both spindled cells and glandular epithelial cells. Muscle marker, for possible mesenchymal differentiation, Desmin was negative. Neuroendocrine markers Synaptophysin and Chromogranin were positive in the solid clusters and trabeculae cells, as well as scattered cells along the glandular epithelium. A Ki67 highlighted an increased mitotic index, compatible with MPNST. Finally, the H3K27me3 was lost in the tumor cells.
The case was compared to the slides from the patient’s previously diagnosed MPNST, approximately 2 years prior, from the right groin. The histology from the prior MPNST case demonstrated a lobular spindled cell proliferation in a fibrous stroma with adjacent lobules of nerve bundles (Figure 2A). Arranged in a storiform pattern the spindled cells were mildly pleomorphic with small to medium, elongated nuclei, vacuolated chromatin, and indistinct cellular borders. Large foci of necrosis were present and frequent mitoses were seen. No divergent differentiation was evident. Immunohistochemical stain S100 was focally positive (Figure 2B). The presenting case tumor of the right arm was diagnosed as a malignant peripheral nerve sheath tumor with extensive heterologous elements, predominantly glandular and mucinous. The histologic features suggested that it was a primary malignancy arising from a pre-existent plexiform neurofibroma, and it did not share any features with the previously diagnosed MPNST to suggest a metastasis (Figure 2C and 2D). Approximately 2-years after diagnosis the patient returned with an excisional biopsy of a left skull mass. Similar to the MPNST of the right arm, histologic features included abundant malignant glandular structures with intervening fascicles of densely packed spindled cells (Figures 2E and 2F). Both histology and immunohistochemical profile were similar, and the lesion was diagnosed as most consistent with metastatic malignant peripheral nerve sheath tumor with abundant heterologous malignant glandular elements.
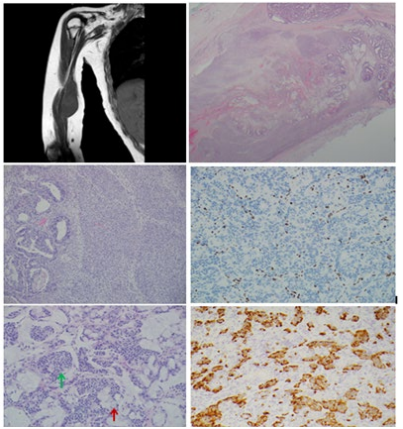
Figure 1: A) MRI of the right upper arm shows an 11.9x3.1x2.6 cm multilobulated hyperintense T2-weighted mass centered in the biceps brachii, coracobrachialis and medial head of the triceps brachii muscles, extending along the median nerve caudally as far as the elbow joint. B) Histologic sections at low magnification show an infiltrative lesion with glandular and spindle cell areas arising in a plexiform neurofibroma. The neurofibromatous elements are appreciated at the periphery of the lesion. C) At higher magnification malignant glandular structures are seen to merge imperceptibly with an equally malignant spindle cell proliferation. The glandular structures are characterized by large cells with scant pale cytoplasm within which are embedded large hyperchromatic nuclei leading to a high nuclear cytoplasmic ratio. The spindle cells are for the most part monotonous with elongated nuclei and scant cytoplasm and arranged in short fascicles. D) By immunohistochemistry, the tumor shows complete loss of expression of H3K27me3. E) Large areas of the tumor showed intestinal type differentiation with an abundance of neuroendocrine cells (green arrow) as well as goblet cells (red arrow), some with a signet ring cell appearance. F) The neuroendocrine-type cells showed strong and diffuse cytoplasmic positivity with chromogranin immunohistochemical stain.

Figure 2: A) The groin lesion resected 2 years prior demonstrates the typical spindle cell morphology of a neurofibroma however mitotic activity is brisk. B) By contrast, the index case shows extensive glandular differentiation and marked tumor necrosis. C) The metastatic lesion to the skull was characterized by a mixture of spindle cells, neuroendocrine cells, glandular structures and even globlet cells reminiscent of signet ring cells (green arrow). D) Clusters of neuroendocrine cells were readily identified in sections from the skull metastasis. These cells were strongly and diffusely positive for synaptophysin and chromogranin.
Discussion
Patients with NF1 represent a population found to be at increased risk for developing MPNST, with reported incidence of 4.6% compared to 0.001% in the general clinic population and a more recently updated population lifetime risk reported at 8-13% [8,10]. Approximately 50% of all MPNSTs arise in the NF1 patient population, and epithelial divergent differentiation often occurs in the context of MPNSTs arising in NF1 patients [5,8]. However, NF1 patients are at a greater risk of developing a variety of different tumors. An increased risk of early onset breast cancer is established and even reports of early onset colon cancers have been reported in NF1 patients [11,12]. The importance in the recognition of the ability of MPNST to develop glandular divergent differentiation is in the risk for misdiagnosis, as no definitive prognostic value has been determined [5]. Based on our review, this case of MPNST is unique as it demonstrates extensive glandular epithelial divergent differentiation, while prior descriptions mention such features as only being discreet or localized [5]. Additionally, while neuroendocrine differentiation has been previously established in association with the divergent glandular epithelium, it has been previously described as neuroendocrine cells within the glandular epithelium resembling an intestinal differentiation, not as the solid nests and trabeculae seen in our case [13]. Fortunately this case was an excisional biopsy and not a core biopsy, so while the glandular component in this case was extensive, there still remained sufficient histologic features for an accurate initial diagnosis.
Disclosure
The authors declare that there are no conflicts of interest regarding the publication of this paper.
References
1. Goldblum JR, Folpe AL, Weiss SW, Enzinger FM, Weiss SW (2014) Enzinger and Weiss’s soft tissue tumors. 6th ed. Philadelphia, PA: Saunders/Elsevier, 1155.
2. James AW, Shurell E, Singh A, Dry SM, Eilber FC (2016) Malignant Peripheral Nerve Sheath Tumor. Surg Oncol Clin N Am 25(4):789-802.
3. Fletcher CDM, World Health Organization (2013) International Agency for Research on Cancer. WHO classification of tumours of soft tissue and bone. 4th ed. Lyon: IARC Press, 468.
4. Ducatman BS, Scheithauer BW (1984) Malignant peripheral nerve sheath tumors with divergent differentiation. Cancer 54(6):1049-1057.
5. Miki Y, Thway K (2017) Malignant Peripheral Nerve Sheath Tumor With Divergent Glandular Differentiation. Int J Surg Pathol 25(4):310-313.
6. Guo A, Liu A, Wei L, Song X (2012) Malignant peripheral nerve sheath tumors: differentiation patterns and immunohistochemical features - a mini-review and our new findings. J Cancer 3:303-309.
7. Zou C, Smith KD, Liu J, Lahat G, Myers S, et al. (2009) Clinical, pathological, and molecular variables predictive of malignant peripheral nerve sheath tumor outcome. Ann Surg 249(6):1014-1022.
8. Ducatman BS, Scheithauer BW, Piepgras DG, Reiman HM, Ilstrup DM (1986) Malignant peripheral nerve sheath tumors. A clinicopathologic study of 120 cases. Cancer 57(10):2006- 2021.
9. Cleven AH, Sannaa GA, Briaire-de Bruijn I, Ingram DR, van de Rijn M, et al. (2016) Loss of H3K27 tri-methylation is a diagnostic marker for malignant peripheral nerve sheath tumors and an indicator for an inferior survival. Mod Pathol 29(6):582-590.
10. Evans DG, Baser ME, McGaughran J, Sharif S, Howard E (2002) Malignant peripheral nerve sheath tumours in neurofibromatosis 1. J Med Genet 39(5):311-314.
11. Anderson JL, Gutmann DH (2015) Neurofibromatosis type 1. Handb Clin Neurol 132:75-86.
12. Shearer P, Parham D, Kovnar E, Kun L, Rao B, et al. (1994) Neurofibromatosis type I and malignancy: review of 32 pediatric cases treated at a single institution. Med Pediatr Oncol 22(2):78-83.
13. Christensen WN, Strong EW, Bains MS, Woodruff JM (1988) Neuroendocrine differentiation in the glandular peripheral nerve sheath tumor. Pathologic distinction from the biphasic synovial sarcoma with glands. Am J Surg Pathol 12(6):417- 426.
Copyright: © 2025 This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.