
Journal of Clinical Review & Case Reports(JCRC)
ISSN: 2573-9565 | DOI: 10.33140/JCRC
Impact Factor: 1.823


Case Report - (2024) Volume 9, Issue 3
Acute Tubular Necrosis–Rare Presentation of Severe Hypercalcemia: A Case Report
2Department of Nephrology and Hypertension,Cleveland Clinic Lerner College of Medicine of Case Wester, USA
USA
#Equally contribution
Received Date: Feb 12, 2024 / Accepted Date: Feb 19, 2024 / Published Date: Mar 01, 2024
Abstract
Renal dysfunction due to hypercalcemia typically manifests with impairment in the ability to concentrate urine, decreased glomerular filtration rate (GFR), natriuresis, nephrocalcinosis and diabetes insipidus. Acute Tubular Necrosis (ATN) is a rare manifestation of severe hypercalcemia (defined as serum calcium greater than 20 mg/dl). We present a case of severe hypercalcemia resulting from Milk-Alkali Syndrome caused by chronic ingestion of calcium-based antacids (TUMS) resulting in Acute Kidney Injury (AKI) from ATN.
Keywords
Hypercalcemia, Acute Tubular Necrosis, Milk-Alkali Syndrome, Calcium-Alkali Syndrome, Acute Kidney Injury
Introduction
ATN is a rare presentation of hypercalcemia. The direct effect of hypercalcemia on renal tubules is not clearly understood and minimal literature is available. The proposed mechanism of ATN from high calcium intake is likely caused by direct vasoconstriction of renal arterioles, polyuria, and diabetes insipidus. The complications associated with ATN are often life threatening with in-hospital survival rate of approximately 50% [1]. The co-existence of severe hypercalcemia and acute renal failure should prompt correlation by the clinician in order to hasten early diagnosis followed by timely treatment to prevent accompanying complications.
Historically, the excessive use of calcium supplements and/ or calcium-based antacids was thought to solely cause MilkAlkali Syndrome (also known as Calcium-Alkali Syndrome). Patients suffering from Milk-Alkali Syndrome present with hypercalcemia, metabolic alkalosis, and renal insufficiency [2]. Renal insufficiency secondary to hypercalcemia from MilkAlkali Syndrome typically manifests with impairment in the ability to concentrate urine, decreased glomerular filtration rate (GFR), nephrocalcinosis and diabetes insipidus.
While hyperparathyroidism and malignancy continue to account for 90% of cases of hypercalcemia, the number of cases of Milk-Alkali Syndrome-induced hypercalcemia and acute kidney injury (AKI) are rising [3,4]. As the use and availability of overthe-counter calcium-based antacids (i.e., TUMS, Rolaids®) become more prevalent and readily accessible in today’s day and age, it is important for clinicians to be aware of AKI and Acute Tubular Necrosis (ATN) as a potential adverse effects of hypercalcemia, especially for patients with pre-existing kidney disease.
ATN is a serious kidney injury defined by the destruction of the epithelial cells lining the renal tubules. Damage to the kidney tubules results in impaired blood flow, in turn oxygenation with subsequent impairment of filtration leading to acute renal failure. Imbalances in electrolytes and waste products lead to elevations in the fractional excretion of sodium (FENa), serum phosphate, serum urea, serum creatinine, and serum potassium. As ATN can be caused by a multitude of factors, it is important to treat ATN based on the underlying problem. As observed in this case, by promptly treating the underlying cause of hypercalcemia which was ATN, a normalization of calcium levels, an improvement in renal function is achievable. This patient experienced a case of severe hypercalcemia (calcium>20 mg/dl) caused by excessive ingestion of calcium-based antacids resulting in AKI.
Case Presentation
A 73-year-old Caucasian female presented with malaise and fatigue of several weeks duration. She also complained of decreased appetite, nausea, abdominal pain with constipation, dizziness and blurred vision. She had a prior history of nephrolithiasis and hypertension along with a history of basal and squamous cell carcinomas as well as pre melanoma that was successfully managed. Her urine output declined to less than 8 ounces per day one week prior to admission. She reported regularly taking 10-12 tablets of TUMS daily over the course of four weeks for indigestion.
Upon admission, the patient had a temperature of 97.5°F, a blood pressure of 134/67 mmHg and a pulse rate of 76 beats per minute. The patient did not show signs of acute distress. Her initial laboratory workup revealed blood urea nitrogen (BUN) of 42 mg/dl, serum creatinine of 2.9 mg/dl (renal function normal prior to this episode), calcium > 20.0 mg/dl, parathyroid hormone (PTH) of 21.3 pg/mL, and a serum chloride of 90 mmol/L. Abdominal Computed Tomography without contrast revealed normal sized kidneys without renal calculi or obstruction. All the other causes of hypercalcemia were ruled out by the initial laboratory workup.
A diagnosis of hypercalcemia due to Milk-Alkali Syndrome was made based on the excessive TUMS intake. Given the severe case of hypercalcemia with resultant AKI, the course of treatment included discontinuing TUMS, administering intravenous normal saline and calcitonin, and starting the patient on shortterm hemodialysis. Serum calcium normalized following several sessions of hemodialysis with resolution of renal insufficiency. The patient was discharged with normal creatinine and calcium. Thus, the patient’s renal function improved significantly following discontinuation of TUMS and several sessions of acute hemodialysis.
Renal biopsy was performed that revealed the following:
• H&E stain demonstrating diffuse acute tubular injury consistent with acute tubular necrosis. (Figure 1(A))
• PAS stain demonstrates a normal appearing glomerulus (Figure 2A). • Hypertensive arteriolonephrosclerosis and arteriolonephrosclerosis with hyalinosis of mild to moderate degree. (Figure 2(B))
• Mild to moderate focal global glomerulosclerosis (Figure 2©)
• Mild focal tubular atrophy and interstitial fibrosis
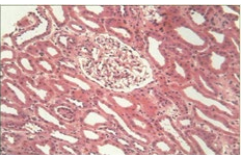
Figure 1A: Acute Tubular Injury.
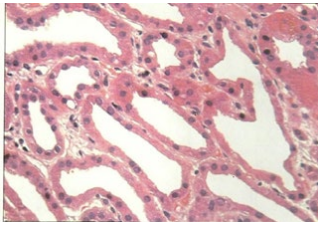
Figure 1B: Tubular Specification.
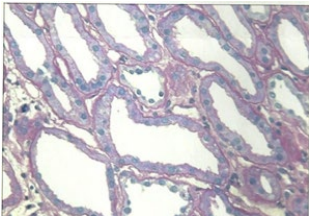
Figure 1C: Reduced Brush Border.
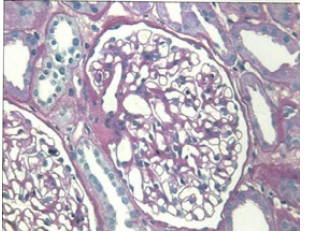
Figure 2A: Normo Cellular Glomerulus.
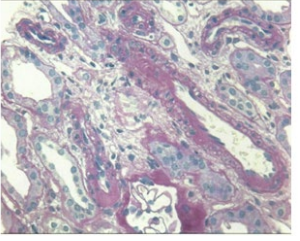
Figure 2B: Ateriosclerosis.
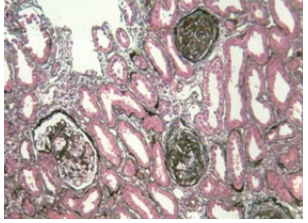
Figure 2C: Focal Glomerulosclerosis.
Discussion
The calcium ion is an essential regulator of several physiological processes including muscle contraction, secretory mechanisms, and neuronal excitation.5 Hypercalcemia can result from multiple causes including malignancies, primary hyperparathyroidism, immobilization, ingestion of vitamins A and D, excessive thiazide diuretic use, granulomatous diseases, chronic renal failure, Milk-Alkali Syndrome, Addison’s Disease, Familial Hypocalciuric Hypercalcemia, and intrinsic bone diseases (e.g., Paget’s disease). 5 The clinical features of hypercalcemia are not specific to the etiology and are shared amongst all potential causes. Patients suffering from hypercalcemia can present with dehydration, nausea, anorexia, vomiting, abdominal pain, and altered mental status. 5 Some of these symptoms were observed in this case. The potential renal consequences of hypercalcemia are afferent arteriole vasoconstriction, decreased tubular sodium reabsorption, nephrogenic diabetes insipidus, pre-renal azotemia, nephrocalcinosis, tubulointerstitial fibrosis, and ATN [6].
While current medical literature supports the hypothesis that hypercalcemia can induce ATN, the mechanism behind this phenomenon has not been clearly defined and a paucity of literature exists in this area. Cytonecrosis of renal tubular cells following elevated serum calcium levels has been documented in both acute as well as in chronic settings [7,8]. In rats given a single three-hour infusion of calcium gluconate, mitochondrial and basement membrane calcification with cell swelling and necrosis were observed when serum calcium exceeded 15 mg/ dl [7,8]. The extent of tubular necrosis correlated with the degree of hypercalcemia such that 5% of renal tubular profiles contained damaged cells for each 1 mg/dl increase in serum calcium above 16 mg/dl [8]. These promising results suggest a stronger relationship between hypercalcemia and ATN.
Currently, there are three known alterations in tubular dynamics that directly contribute to the development of ATN: obstruction, back-leak, and activation of tubuloglomerular feedback [9,10]. In this patient, obstruction was not observed as evidenced by absence on CT imaging. In this case, we see that just as the findings of recent human studies [9,11] suggest, obstruction alone is unlikely to account for the profound dysfunction in clinical ATN. In these studies, it has been found that forced dieresis in humans using furosemide [12] or mannitol [13] did not impact the survival and renal recovery rate of patients with established acute renal failure.
There are several mechanisms proposed through which hypercalcemia can contribute to the development of ATN. The direct vasoconstriction effects of excess calcium ions on afferent arteriolar smooth muscle cells, activation of tubuloglomerular feedback in ATN dysfunction, and the symptoms of polyuria and nephrogenic diabetes insipidus caused by hypercalcemia are all proposed to contribute largely towards the renal injury observed in clinical ATN [6,9,14,15]. An increase in renal vascular resistance with a reduction in renal blood flow appears to result from direct vasoconstriction effects of excess calcium ions on the smooth muscle cells of the afferent arterioles [6,7]. Hypercalcemia inhibits luminal Na-K ATPase causing increased sodium load in the renal tubules. By activating the calciumsensing receptor in the medullary thick ascending limb, the Na and K cotransport is inhibited. This causes natriuresis and a depletion of blood volume [17]. The resulting increased delivery of sodium chloride to the macula densa due to cellular abnormalities in the ischemic proximal tubule would be expected to induce afferent arteriolar constriction via A1 adenosine receptor activation. Ultimately, the excess of calcium causes a combination of a depleted blood volume and afferent arteriolar constriction leading to a decreased GFR and an impaired renal function [9].
The workup, including serum studies, urinalysis, biopsy, and CT scan of the abdomen, is critically important for identifying the causes of hypercalcemia and determining whether the resulting AKI is caused by pre-renal, renal, or post-renal acute renal injury. Specifically, understanding the serum BUN, serum creatinine levels, BUN to creatinine ratio, chloride, creatinine, and sodium levels, helped in identifying the cause and nature of the patient’s hypercalcemia and acute kidney injury. In this patient, physical examination and vital signs on presentation were also suggestive of dehydration. The decreased BUN to creatinine ratio and presence of oliguria are suggestive of ATN. Hypercalcemia can cause ATN from direct effect on tubules, other than ischemic ATN from polyuria, nephrogenic diabetes insipidus, and pre-renal vasoconstriction [16].
Treatment of acute hypercalcemic nephropathy should start with identifying and correcting the underlying cause. In some cases, this can mean restoring intravascular fluid volume with intravenous fluids, using short-term hemodialysis, and/or interrupting bone resorption using bisphosphonates, calcitonin, or mithramycin. Increasing urine output by using saline diuresis and loop diuretics can be used to manage the fluid volume and lower the serum calcium. Though commonly used, studies have found little-to-no evidence supporting the use of loop diuretics in improving patient outcomes in AKI. Some benefits of using loop diuretics include converting oliguric into non-oliguric AKI, managing fluid overload, and inhibiting prostaglandin dehydrogenase to improve renal blood flow. However, this technique has an ability to aggravate AKI by decreasing effective circulating blood volume, increasing the aggregation of Tamm-Horsfall protein, and creating electrolyte abnormalities and metabolic alkalosis [18-20]. If hypercalcemia is promptly reversed, eventual recovery of nephron function can be expected. However, if hypercalcemia remains chronic and persistent, various degrees of interstitial calcification, inflammation, and sclerosis have the potential of evolving. As the duration of hypercalcemia increases, the less reversible its effects become. By waiting too long, nephrocalcinosis and chronic renal insufficiency can result [7].
Ultimately, the combination of dehydration and Milk-Alkali Syndrome resulted in severe hypercalcemia. By the mechanisms proposed above, it is likely that the lower effective circulating blood volume, disruption of tubuloglomerular feedback, and afferent arteriole vasoconstriction led to the development of ATN marked by oliguria, decreased GFR and a elevated BUN and creatinine.
Conclusions
The coexistence of ATN and severe hypercalcemia from MilkAlkali Syndrome in this patient suggests that there is a deeper connection between these two conditions. The complications associated with ATN are often life threatening with in-hospital survival rate of approximately 50%. Any case presenting with hypercalcemia and acute renal failure should be investigated for ATN for early diagnosis and should receive timely treatment.
Consent
“Written informed consent was obtained from the patient for publication of this case report and accompanying images. A copy of the written consent is available for review by the Editorin-Chief of this journal.”
Statement of Ethics
The authors have no statement to disclose for this publication.
Conflict of Interest Statement
The authors declare that there are no conflicts of interest regarding this publication.
Funding Sources
The authors did not obtain funding for this manuscript.
References
1. Mutnuri, S., Agraharkar, M., Lerma, E. (2021). Acute Tubular Necrosis: Practice Essentials, Pathophysiology, Etiology. Medscape.com. Published October 17, 2021. Accessed April 22, 2022.
2. Zayed, R.F., Millhouse, P.W., Kamyab, F., Ortiz, J.F., & Atoot, A. (2021). Calcium-Alkali Syndrome: Historical Review, Pathophysiology and Post-Modern Update. Cureus, 13(2), e13291.
3. Watson, S.C., Dellinger, B.B., Jennings, K., & Scott, L.A. (2012). Antacids, altered mental status, and milk-alkali syndrome. Case reports in emergency medicine, 2012, 942452.
4. Parker, B.M. (2021). Calcium Alkali Syndrome Treated With Hemodialysis. Cureus, 13(3), e13749.
5. Moyses-Neto, M. (2006). Acute renal failure and hypercalcemia. Ren Fail, 28, 153-159.
6. Mahnensmith, R,L. (1992). Vignette in clinical pathophysiology. Am J Kid Dis, 29, 604-608.
7. Epstein, F.H. (1968). Calcium and the kidney. Am J Med, 45, 700-714.
8. Ganote, C.E., Philipsborne, D.S., & Chen, E. (1975). Acute calcium nephrotoxicity. Arch Pathol, 99, 650-657.
9. Devarajan, P. (2005). Cellular and molecular derangements in acute tubular necrosis. Current Opinions in Pediatrics, 17(2), 193-199.
10. Schrier, R.W., Wang, W., Poole, B., et al. (2005). Acute renal failure: definitions, diagnosis, pathogenesis, and therapy. J Clin Invest, 114, 5-14.
11. Wangsiripaisan, A., Gengaro, P.E., Edelstein, C.L., et al. (2001). Role of polymeric Tamm-Horsfall protein in cast formation: oligosaccharide and tubular fluid ions. Kidney Int, 59, 932-940
12. Cantarovich, F., Rangoonwala, B., & Lorenz, H (2004). High dose furosemide for established ARF: a prospective randomized, double blind, placebo controlled, multi-center trial.Am J Kidney, 44, 402-409.
13. Gambaro, G., Bertaglia, G., Puma, G., et al. (2002). Diuretics and dopamine for the prevention and treatment of acute renal failure: a critical reappraisal. J Nephrol, 15, 213-219.
14. Kwon, O., Corrigan, G., Myers, B.D. (1999). Sodium reabsorption and distribution of Na/K ATPase during post ischemic injury to the renal allograft. Kidney Int, 55, 963- 975.
15. Goldfarb, S., Agus, Z.S. (1984). Mechanism of the polyuria of hypercalcemia.Am J Nephrol, 4, 69-76.
16. Lins, L.E. (1978). Reversible renal failure caused by hypercalcemia. Acta Med Scand, 203, 309-314.
17. Benabe, J.E., Martinez-Maldonado, M. (1978). Hypercalcemic nephropathy. Arch Intern Med, 138, 777- 779.
18. Hegde, A. (2020). Diuretics in Acute Kidney Injury. Indian J Crit Care Med, 24(Suppl 3), S98-S99.
19. Hanif, M.O., Bali, A., & Ramphul, K. (2022). Acute Renal Tubular Necrosis. (Updated 2021 Oct 12). In: StatPearls (Internet). Treasure Island (FL): StatPearls Publishing; 2022 Jan.
20. Senor, B. (2022). Acute Kidney Injury from Acute Tubular Necrosis vs Prerenal Cause-How Do I Tell the Difference? Enjoin. Published February 4, 2021. Accessed May 1, 2022.
Copyright: © 2025 This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.